О журнале
Рекомендации
Использование облачной инфраструктуры для анализа данных секвенирования микроРНК
А.В. Кураченко, И.В. Заигрин, Ф.С. Шарко, А.Б. Теслюк, А.В. Недолужко
для анализа данных секвенирования микроРНК
_______________________
А.В. Кураченко, И.В. Заигрин, Ф.С. Шарко, А.Б. Теслюк, А.В. Недолужко
Аннотация
Секвенирование с использованием технологий next-generation sequencing (NGS) позволяет получать значительное число информации о геноме или транскриптоме конкретного организма, однако дальнейшая обработка этих данных не всегда тривиальна.
В данной работе мы попытались реализовать базовый алгоритм, который на основе существующих общедоступных и разработанных нами программ и скриптов позволит обрабатывать данные секвенирования микроРНК у различных организмов. В качестве среды для проведения обработки данных использована платформа Galaxy, которая была установлена на виртуальный кластер под управлением Torque с планировщиком заданий Maui на базе вычислительного облака OpenStack.
Ключевые слова: микроРНК, секвенирование, NGS, платформа Galaxy, веб-инструмент.
Секвенирование – это процесс, направленный на определение последовательности нуклеотидов – аденина, тимина (урацила), гуанина и цитозина – в молекулах ДНК или РНК. Разнообразие последовательностей генов и некодирующих участков генома обеспечивает колоссальные различия между видами, а также внутривидовую изменчивость. В последние годы секвенирование стало рутинной процедурой, стоимость которой продолжает снижаться. Особый прогресс в области расшифровки геномов наметился после внедрения технологий секвенирования последнего поколения (next-generation sequencing), благодаря которым появилась возможность производить терабайты генетических данных [1].
После публикации первого генома человека секвенирование ДНК превратилось в один из повседневных инструментов молекулярной биологии [2]. Одним из перспективных направлений геномики стало изучение функционирования генома, в частности, исследование регуляции работы генов.
Изначально работы по анализу данных секвенирования сводились к формированию списка заданий и последовательному использованию большого количества программ/скриптов. На изменение параметров в промежуточных задачах тратилось много времени. Отсутствие удобного пользовательского интерфейса превращало биоинформатическую обработку в длительную и не всегда простую процедуру.
За последние пять лет появилось значительное число облачных сервисов, разработанных для анализа данных секвенирования. Большая их часть, являясь довольно гибкими инструментами, отличается высокой коммерческой стоимостью, бесплатные же аналоги предоставляют недостаточные возможности для построения алгоритмов задач, а также имеют ограничения на входные объемы анализируемых данных (задач) – Discovery Net [3], Galaxy [4], Taverna [5, 6], Kepler [7], Triana [8], Conveyor [9], Pegasus [10] и другие.
В данной работе мы представляем инструментальную среду для анализа данных секвенирования молекул микроРНК. Известно, что такие молекулы (18–25 пар нуклеотидов) принимают непосредственное участие в регуляции экспрессии генов и влияют на многие процессы в клетках, регулируя рост и развитие тканей, дифференцировку клеток, клеточный ответ на абиотические и биотические факторы [11]. Кроме того, молекулы микроРНК могут стать одним из маркеров заболеваний, в том числе онкологических и инфекционных [12,13]. Здесь речь пойдет о методе секвенирования молекул микроРНК с помощью секвенаторов SOLID, биоинформатических методах фильтрации и обработки данных секвенирования, а также об интеграции методов обработки данных в облачные сервисы с использованием интуитивно понятного веб-интерфейса.
Методы
Секвенирование фракции микроРНК в транскриптоме клеток. Тестовые данные для разработки интерфейса были получены с использованием геномного секвенатора SOLiD 4.0, установленного в лаборатории геномики НИЦ «Курчатовский институт». Для этого нами были отсеквенированы молекулы микроРНК, выделенные из трёх типов тканей хризантемы садовой (Chrysanthemum morifolium): вегетативная ткань, ткань растения на стадии бутонизации и ткани соцветия. Из фракции микроРНК каждой ткани были созданы кДНК-библиотеки – молекулы микроРНК, переведённые в двуцепочечную форму с прикреплёнными (используя фермент лигазу) по концам ДНК-адаптерами, необходимыми для амплификации и секвенирования. Необработанные данные секвенирования в форматах *.csfasta и *.qual являлись отправной точкой для нашей работы.
Файл *.csfasta (color space fasta) содержит информацию о считанных последовательностях в цветовом формате (где каждому цвету соответствуют два нуклеотида) и их координатах. В *.qual файле представлена информация о качестве данных секвенирования.
Технология SOLiD 4.0 основана на циклическом лигазном секвенировании, в каждом цикле которого фермент лигаза пришивает к 5’-концу ДНК-библиотеки флуоресцентномеченый олигонуклеотид. После идентификации флуоресцентной метки проводится её отщепление и регенерация субстратного комплекса, удлинённого на 5 нуклеотидов. Считывание последовательности проводится с помощью фиксации цветовых сигналов, которые кодируются цифрами – 0 (синий), 1 (зеленый), 2 (желтый) и 3 (красный). Каждый из этих сигналов определяет два соседних нуклеотида. Например, динуклеотид TA (тимин–аденин) соответствует цифре 3 (табл. 1).

Табл. 1 Система динуклеотидного кодирования, используемая в секвенаторах SOLiD
Биоинформатический анализ данных микроРНК. Система динуклеотидного кодирования, которая используется в технологии секвенирования, накладывает ограничения на последующую обработку данных. Одна ошибка в «прочтении» может полностью изменить дальнейшую последовательность нуклеотидов в секвенируемой молекуле и привести к неверным выводам. Поэтому при первичном анализе необходимо выбирать последовательности, обладающие высоким уровнем качества прочтения последовательности, которое представлено в файле *.qual. Следует отметить, что файл *.qual, генерируемый секвенатором, представляет собой не что иное, как значения шумов, фиксируемых при считывании цветовой метки. Для получения достоверных результатов нами проводилась фильтрация данных по качеству, использовались только чтения с QV (quality value) > 15. Кроме этого, с помощью программы Cutadapt из данных исключались последовательности адаптеров, использовавшихся при создании кДНК-библиотек [14].
В дальнейшем проводилось картирование отфильтрованных чтений на базу данных микроРНК – MirBase 19.0 [15]. Для выравнивания на известные микроРНК использовалась программа Bowtie, которая производит картирование несколькими способами, а также эффективно распараллеливает вычисления, что позволяет значительно увеличить скорость и качество анализа по сравнению с аналогичными программами [16].
Перенос скриптов (утилит) для обработки данных микроРНК-анализа на платформу Galaxy. Galaxy – открытая веб-платформа, разработанная специально для геномных исследований, с помощью которой можно интегрировать различные программные пакеты для обработки и анализа данных в единую систему с удобным веб-интерфейсом. Основные объекты Galaxy: история (набор всех входных, промежуточных и выходных данных), утилиты (набор операций, позволяющих производить разнообразную обработку данных), последовательность выполняемых действий (поток операций, представляющий все этапы анализа данных).
На веб-платформе Galaxy представлен большой набор утилит, однако они рассчитаны на обработку геномных данных, в то время как при анализе микроРНК имеются свои особенности, из-за чего встроенные утилиты не всегда эффективны, Для самостоятельной работы в Galaxy реализован простой способ добавления собственных утилит (рис. 1).
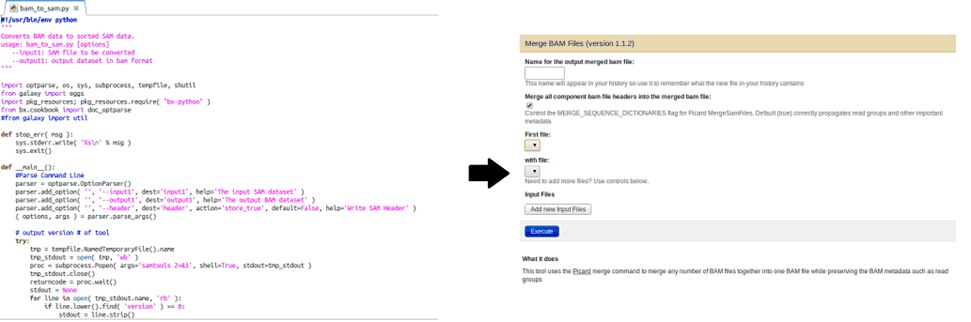
Рис. 1. Окно запуска скрипта (утилиты) в Galaxy
Создание утилиты, по сути, заключается в написание скрипта на одном из популярных языков программирования (Python, Perl, Shell, С++) и конфигурационного файла к нему. Нами были добавлены необходимые утилиты, позволившие реализовать схему обработки данных микроРНК в Galaxy. Последовательность выполняемых действий (workflow) в Galaxy представляет собой поток утилит, настроенных на последовательное выполнение (рис. 2). К достоинствам платформы следует отнести удобный графический редактор.
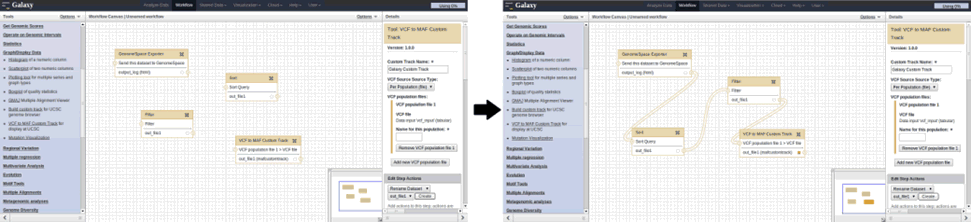
Рис. 2. Создание workflow в Galaxy (добавление утилит и связывание их)
Мы реализовали несколько алгоритмов, которые принимали исходные данные для последующей их фильтрации и картирования, выдавая в конечном результате списки обнаруженных семейств микроРНК, графики и таблицы для наглядной демонстрации полученных результатов.
Архитектура сервисов по обработке данных секвенирования. После отработки алгоритмов анализа данных и их реализации в системе Galaxy возник вопрос о создании сервиса, который позволит другим исследователям пользоваться нашим инструментарием. В качестве архитектуры для создания сервиса был выбран виртуальный кластер на базе системы организации облачных вычислений Openstack. Облачные технологии хорошо подходят для подобных задач, поскольку позволяют гибко регулировать вычислительные мощности и размер системы хранения. При поступлении экспериментальных данных запускается необходимое количество виртуальных узлов кластера для их обработки, после завершения анализа выделенные ресурсы высвобождаются. Таким путем идут многие биоинформационные компании, когда строят свои сервисы на базе облаков Amazon или Rackspace.
Рассмотрим более подробно компоненты, из которых состоит разработанная система.
● OpenStack – программное обеспечение с открытым исходным кодом для построения публичных и частных облачных установок. Включает в себя подсистемы OpenStack Compute (Nova – предоставляет запуск виртуальных машин по запросу, аналогичен Rackspace Cloud Servers или Amazon EC2), OpenStack Object Storage (Swift – обеспечивает хранение блобов или объектов, примерный аналог Rackspace Cloud Files или Amazon S3) и Openstack Image Service (Glance – обеспечивает обнаружение, хранение и получение образов виртуальных машин для Nova). OpenStack осуществляет контроль и обслуживание вычислительных узлов. Вычислительный узел представляет собой образ виртуальный машины, готовый к запуску с необходимыми параметрам (сетевой адрес, название и др.). Запуск дополнительных вычислительных узлов и выключение (для освобождения вычислительных ресурсов облака) осуществляется по запросу балансировщика.
● TORQUE – менеджер распределенных ресурсов для вычислительных кластеров из машин под управлением Linux и других Unix-подобных операционных систем, одна из современных версий Portable Batch System (Система пакетной обработки заданий). Распространяется под свободной лицензией OpenPBS Software License.
Основная функция TORQUE – распределение вычислительных задач среди доступных вычислительных ресурсов. Задания создаются пользователем через Galaxy и поступают в очередь задач на сервере заданий (Master node). Вычислительные узлы (worker node) взаимодействуют с сервером заданий, получая задания на выполнение планировщиком (Maui).
● Maui (Maui cluster scheduler) – планировщик заданий в параллельных и распределенных вычислительных системах (кластерах). Позволяет выбирать различные политики планирования, поддерживает динамическое изменение приоритетов, исключения. Планировщик взаимодействует с клиентом заданий (Job Executor) вычислительного узла, запрашивая информацию о состоянии системных ресурсов, а также с сервером заданий для получения списка заданий, доступных для выполнения. Он определяет, когда данное задание будет запущено и какие ресурсы ему будут выделены.
● Balancer – программный компонент, написанный на языке Python, контролирующий нагрузку виртуального кластера. Контролируя поступающие задачи в очередь задач Torque (от какого пользователя поступило задание, количество заданий, количество занятых вычислительных узлов), балансировщик может послать запрос к OpenStack Nova на запуск (выключение) дополнительных вычислительных узлов.
В виртуальном кластере изначально определено максимальное количество вычислительных узлов. Выключенные вычислительные узлы определены состоянием «недоступен».
Обработка данных микроРНК-анализа с помощью облачной платформы. Для начала работы с системой пользователю необходимо зарегистрироваться (регистрация в системе бесплатна). Далее на главной странице следует использовать пункт «Добавление файла» и загрузить данные микроРНК на сервер (в настоящее время существует возможность работы только с файлами секвенаторов SOLiD). Затем можно выбрать один из интересующих пунктов:
● простой анализ данных (происходит поиск только по известным микроРНК),
● полный анализ данных (происходит поиск по микроРНК, пре-микроРНК) (рис. 3).
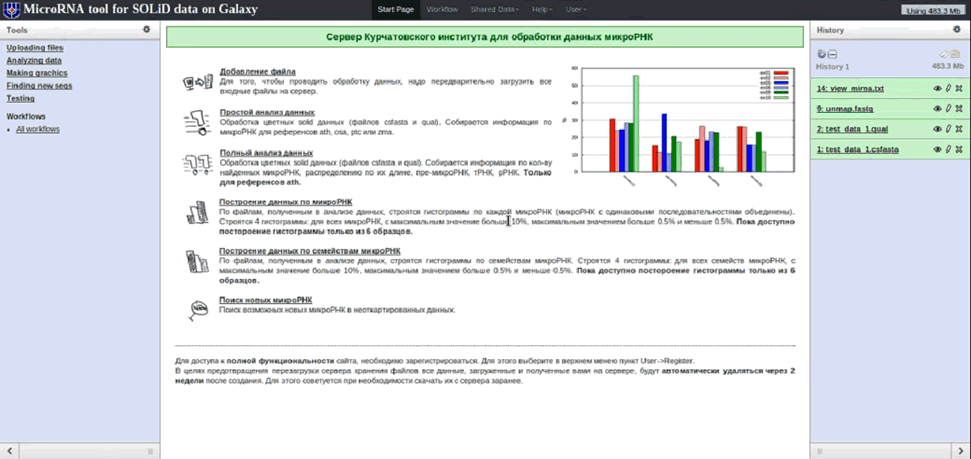
Рис. 3. Главная страница платформы
После выбора этих пунктов пользователь перенаправляется на страницу запуска алгоритмов. Здесь он сможет выбрать загруженные на сервер данные, а также изменить параметры фильтрации и картирования данных (рис. 4).
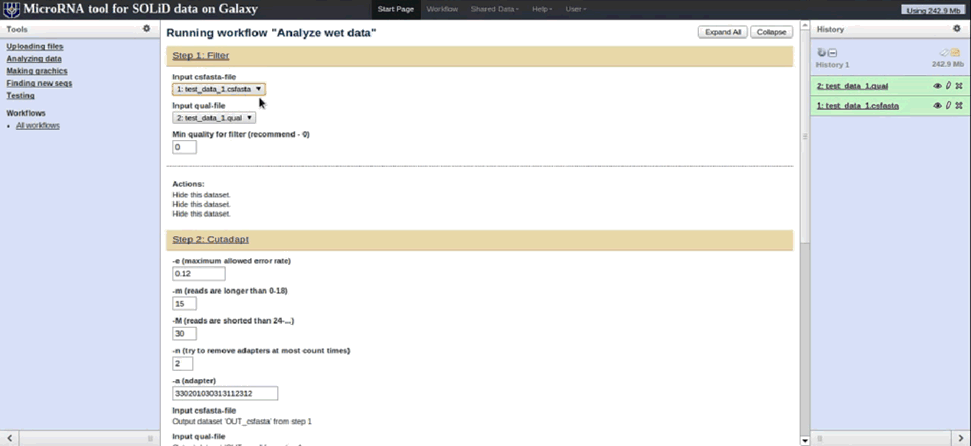
Рис. 4. Окно запуска workflow для анализа геномных данных
По окончании анализа пользователь получит данные о встречающихся последовательностях микроРНК. С помощью других алгоритмов, ссылки на которые также находятся на главной странице, можно:
● построить графики и таблицы для наглядного отображения данных,
● найти в неоткартированных данных последовательности, которые могут являться потенциально новыми для науки семействами или типами микроРНК (рис. 5).
Все данные доступны для просмотра в веб-интерфейсе и могут быть загружены на личный компьютер.
Рис. 5. Просмотр результатов в виде списка и графика, демонстрирующего встречаемость найденных микроРНК
Результаты. Для проверки работы предложенной нами инструментальной среды использовались данные секвенирования молекул микроРНК из трех типов тканей хризантемы садовой (Chrysanthemum morifolium) – вегетативная ткань, ткань растения на стадии бутонизации и ткани соцветия. Три типа сконструированных кДНК-библиотек микроРНК хризантемы садовой были отсеквенированы с использованием геномного секвенатора ABI SOLiD 4.0. Полученные данные были обработаны с помощью разработанного алгоритма (доступен по адресу: bio.nanocloud.su), лишь малая часть из них картировалась на известные микроРНК A. thaliana (табл. 2), что, по-видимому, связано с филогенетическим положением изучаемого вида.
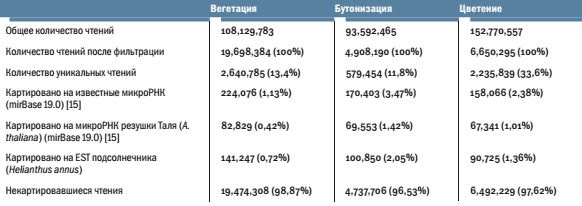
Таблица 2. Анализ данных геномного секвенирования с использованием разработанной инструментальной среды на базе облачных технологий
Картирование чтений, сгенерированных на секвенаторе, на базу известных науке микроРНК показало, что в транскриптоме хризантемы представлено несколько семейств консервативных микроРНК, причем пять из них доминируют над остальными: miR159, miR164, miR166, miR167 и miR396. Особый интерес представляют недоминирующие консервативные микроРНК, чья экспрессия во многом зависит от типа ткани, из которой они выделены. Были выявлены микроРНК, представленность которых значительно отличается в зависимости от физиологии органов растений (табл. 3).
Отличия в экспрессии микроРНК в разных тканях, по-видимому, связаны с физиологическими процессами, протекающими в клетках. Например, молекулы из семейства miR162 и miR168 по механизму обратной связи репрессируют экспрессию генов DICER-LIKE 1 (DCL1), которые отвечают за синтез белков, регулирующих созревание самих микроРНК [17]; микроРНК из семейства miR156 регулируют переход растения из ювенильной в генеративную стадию [18,19], а miR157, вероятно, влияют на синтез DEAD-box РНК-хеликазы [17].

Таблица 3. Консервативные микроРНК, уровень экспрессии которых существенно различается в зависимости от типа ткани (уровень экспрессии представлен в процентах)
Для слабоизученных видов, геномы которых еще неотсеквенированы, особый интерес представляет поиск потенциально новых молекул микроРНК. В ходе вторичного анализа в массиве данных была выявлена последовательность потенциально новой микроРНК (UAUGAAGGUAGUCUAGCCCAC), которая обнаружена также в эксперименте, направленном на изучение консервативных микроРНК в транскриптоме артишока (Cynara sp.) [20].
Таким образом, нами была разработана инструментальная среда, которая позволяет обрабатывать данные секвенирования микроРНК. В настоящий момент она доступна всем желающим по адресу bio.nanocloud.su. В дальнейшем планируется расширить возможности системы для обработки данных полногеномного секвенирования.
Работа поддержана: Российским фондом фундаментальных исследований (РФФИ, грант 12-07-31039 мол_а), стипендией Президента Российской Федерации (СП-2056.2012.5), Фондом содействия развитию малых форм предприятий в научно-технической сфере (программа УМНИК).
Литература
1. Schuster S.C. Next-generation sequencing transforms today's biology // Nature Methods. 2008. №5 (1). P. 16–18.
2. Lander E.S. et al. Initial sequencing and analysis of the human genome // Nature. 2001. № 409 (6822). P. 860–921.
3. Rowe A., Kalaitzopoulos D., Osmond M., Ghanem M., Guo Y. The discovery net system for high throughput bioinformatics // Bioinformatics. 2003. № 19(1). P. i225–i231.
4. Giardine B., Riemer C., Hardison R.C. et al. Galaxy: a platform for interactive large-scale genome analysis // Genome Research. 2005. № 15(10). P. 1451–1455.
5. Oinn T., Addis M., Ferris J. et al. Taverna: a tool for the composition and enactment of bioinformatics workflows // Bioinformatics. 2004. № 20(17). P. 3045–3054.
6. Hull D., Wolstencroft K., Stevens R. et al. Taverna: a tool for building and running workflows of services // Nucleic Acids Research. № 34 (Web Server issue). W. 729–32.
7. Ludäscher B., Altintas I., Berkley C. et al. Scientific Workflow Management and the Kepler System. Special Issue: Workflow in Grid Systems // Concurrency and Computation: Practice & Experience. 2006. № 18(10). P. 1039–1065.
8. Churches D., Gombas G., Harrison A. et al. Programming scientific and distributed workflow with Triana services // Concurrency and Computation: Practice & Experience. 2006. № 18. P.1021–1037.
9. Linke B., Giegerich R., Goesmann A. Conveyor: a workflow engine for bioinformatic analyses // Bioinformatics. 2011. № 27(7). P. 903–911.
10. Deelman E., Blythe J., Gil Y. et al. Mapping complex scientific workflows onto distributed systems // Journal of Grid Computing. 2003. № 1. P. 25–39.
11. Zhang B., Pan X., Cobb G.P., Anderson T.A. Plant microRNA: a small regulatory molecule with big impact // Developmental Biology. 2006. № 289(1). P. 3–16.
12. Wang F., Sun G.P., Zou Y.F. et al. MicroRNAs as promising biomarkers for gastric cancer // Cancer Biomarkers. 2012. № 11(6). P. 259–267.
13. Wang Y., Brahmakshatriya V., Lupiani B. et al. Integrated analysis of microRNA expression and mRNA transcriptome in lungs of avian influenza virus infected broilers // BMC Genomics. 2012. № 13 (278). doi:10.1186/1471-2164-13-278.
14. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads // EMBnet.journal. 2011. № 17(1). P. 11–12.
15. Griffiths-Jones S., Saini H.K., van Dongen S., Enright A.J. miRBase: tools for microRNA genomics // Nucleic Acids Research. 2008. № 36. P. 154–158.
16. Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome // Genome Biology. 2009. № 10(3). R. 25.
17. Bartel B., Bartel D.P. MicroRNAs: at the root of plant development? // Plant Physiology. 2003. № 132(2). P. 709–717.
18. Poethig R.S. The past, present, and future of vegetative phase change // Plant Physiology. 2010. № 154(2). P. 541–544.
19. Dong Z., Danilevskaya O., Abadie T. et al. A gene regulatory network model for floral transition of the shoot apex in maize and its dynamic modeling // PLoS ONE. 2012. № 7(8). doi:10.1371/journal.pone.0043450.
20. Catalano D., Pignone D., Sonnante G., Finetti-Sialer M.M. In-silico and in-vivo analyses of EST databases unveil conserved miRNAs from Carthamus tinctorius and Cynara cardunculus // BMC Bioinformatics. 2012. № 13. doi:10.1186/1471-2105-13-S4-S12.
_____________________________________
Кураченко Анатолий Викторович - аспирант ФГБУ «Национальный исследовательский центр «Курчатовский институт», инженер-исследователь лаборатории информационной интеграции Курчатовского НБИКС-Центра
Заигрин Игорь Владимирович - студент НИУ «Московский физико-технический
Институт», лаборант-исследователь в ФГБУ «Национальный исследовательский центр «Курчатовский институт»
Шарко Федор Сергеевич - студент НИУ «Московский физико-технический институт», лаборант-исследователь в ФГБУ «Национальный исследовательский центр «Курчатовский институт»
Недолужко Артем Валерьевич - кандидат биологических наук, научный сотрудник ФГБУ “Национальный исследовательский центр «Курчатовский институт»
Теслюк Антон Борисович - научный сотрудник ФГБУ «Национальный исследовательский центр «Курчатовский институт»
© Информационное общество, 2013 вып. 1-2, с. 26-38.